Research on Methodology
In the area of Synthetic Methodology, the Clive Group has made a number of contributions that have withstood the test of time and have become common reactions that are widely used in synthesis. These contributions mainly involve the invention of selenium and tellurium reagents, and the development of radical cyclization, which is an area that has become an extremely large and valuable part of Synthetic Organic Chemistry.
Selenoxide Elimination
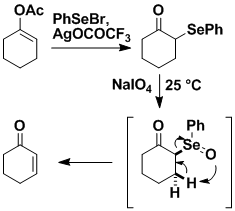
We played a large role in the discovery of organoselenium reagents through the development of selenoxide elimination for generating double bonds, and exploration of the class of reactions we call cyclofunctionalization (see below). Selenoxide elimination is an exceptionally valuable method for introducing double bonds, and it is now such a fundamental part of organic chemistry that it has found its way into standard beginning textbooks.
Cyclofunctionalization
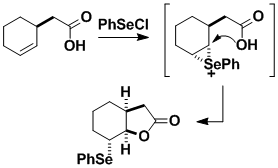
Selenium-induced ring closures serve not only to bring about a cyclization but also to introduce a functional group — hence the name cyclofunctionalization. The method can be used to prepare a wide range of heterocycles, especially those containing O, N, and S. The formation of carbon-carbon bonds is also possible.
Tellurium Reagents
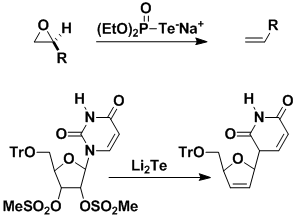
We introduced the first organic reagent containing tellurium [(EtO)2P(O)Te-Na+], which deoxygenates epoxides. The reagent generates a three-membered ring containing tellurium; such species spontaneously expel tellurium to afford an alkene. Based on our understanding of the mechanism, we found that Li2Te converts nucleoside dimesylates into unsaturated nucleosides, which are intermediates in the synthesis of anti-AIDS drugs. Our method has been patented.
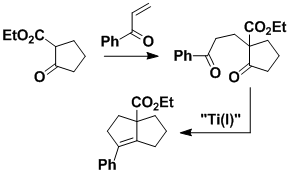
Titanium Reagent for Ring Formation
During the synthesis of compactin and mevinolin we discovered a titanium reagent [formally Ti(I)] that joins carbonyl groups in circumstances where other reagents do not work. The new reagent is very useful for construction of polycyclic compounds; the steps involved are usually straightforward, and sensitive functionality is tolerated.
Radical Cyclization
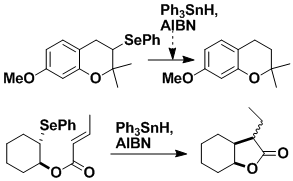
My group has played a very prominent role in the development of radical cyclization, especially through the discovery that phenyl selenides (PhSeR) can be converted into radicals R• by reaction with Ph3SnH and an initiator, such as AIBN. These radicals can then be used for simple reduction or for other purposes, such as cyclization. The PhSe group is now used in most laboratories as one of the standard precursors for radicals in many types of reaction, and is employed across the board — from the construction of small molecules to complex natural products.
Advantages of the PhSe Group
A special feature of the PhSe group as a source of carbon radicals is that it is able to withstand a very wide range of conditions. It can even tolerate the presence of strong base, so that enolate chemistry can be done on substances containing this radical precursor. The usefulness of PhSe has recently been extended by our discovery that it is compatible with the Grubbs catalyst, thus allowing efficient synthesis of polycyclic structures.
New Tin Hydride Reagent
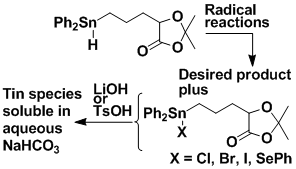
Carbon radicals are usually generated from phenyl selenides or bromides by the action of Ph3SnH or Bu3SnH. Although these methods are extremely popular, some effort is usually needed to separate the reaction product from the tin-containing byproducts. We have prepared a modified stannane which greatly facilitates product isolation because the tin-containing byproducts can be made soluble in aqueous sodium bicarbonate, and then simply extracted into the aqueous layer.
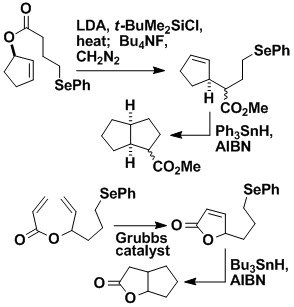
Iterative Ring Formation
We have developed an iterative process for fusing rings onto existing rings. Each new ring can be fused syn or anti to the previous ring. The rings can be added in a linear manner, as shown, or in a clockwise or anticlockwise direction. We have used the linear version of this methodology in the total synthesis of ceratopicanol.
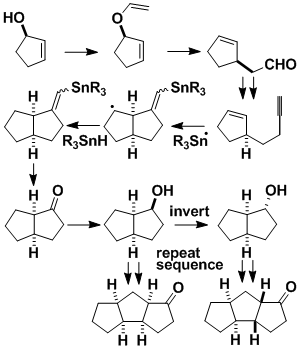
Radical "Bouncing Reactions"
The radical cascade summarized in the Scheme represents a way of carrying out "5-endo-trigonal" cyclizations. These are normally disfavored, but the Scheme shows how to carry out such processes, and we have used the method to synthesize juruenolide C. The initial radical undergoes cyclization, intramolecular hydrogen transfer, and 5-endo-trigonal cyclization. The resulting radical is then reduced by R'3SnH or expels R'3Sn•. Several bonds are formed in a single step, and the stereochemical outcome is controlled by the stereochemistry at the starred atom in the starting material.
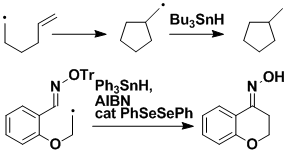
Radical Cyclization with Retention of Functionality
In classical radical cyclizations onto a double bond, functionality is lost as the double bond is replaced by a single bond, and so opportunities for further modifications are not available. We can avoid this restriction by using an O-trityl oxime as the double bond radical acceptor; the product retains a double bond.
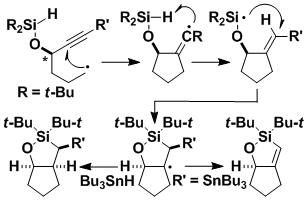
Intramolecular Transfer of Aryl Groups
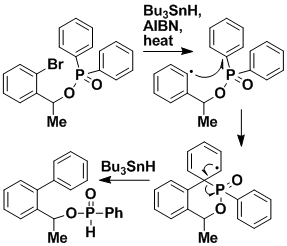
Intramolecular transfer of aryl groups offers a route to important biaryls. We have found a new method for using radical reactions to effect such transfers, using readily available phosphinates.
Radical Cyclization onto an Aromatic Ring
Radical cyclization onto a benzenoid ring has significant potential in synthesis but has not previously been a workable process. We have developed a general method that makes such cyclizations very easy. Our approach is to temporarily convert the benzene ring into a non-aromatic ring that can undergo radical cyclization in the normal way. After the cyclization, the benzene ring is regenerated. The method has been extended to nitrogen heterocycles.
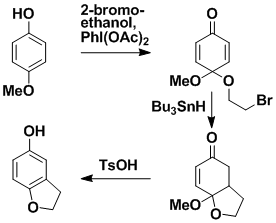
Formation of Benzene Rings
Esters, acids and acid chlorides can be converted via the intermediacy of their corresponding Weinreb amides into benzene derivatives that incorporate the original carbonyl carbon as part of the benzene ring. The process involves treatment of the Weinreb amides with 3-butenylmagnesium bromide and an allylic Grignard reagent, followed by the three simple steps of ring closing metathesis, dehydration and dehydrogenation.
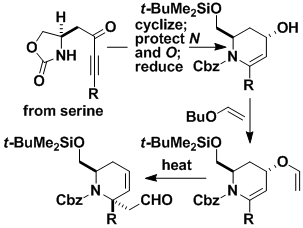
Formation of Optically Active Amines with Quaternary Centers
Optically pure serine can easily be elaborated into functionalized piperidines with a quaternary center next to nitrogen. Such compounds can be processed to afford many optically pure nitrogen-containing substances of pharmaceutical relevance.
Intramolecular Conjugate Displacement (ICD)
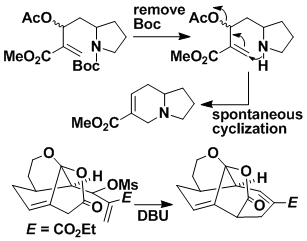
The ICD reaction is so-named because it combines the attributes of a intramolecular conjugate addition and an SN2' displacement. The reaction occurs much more readily than a normal Michael addition. The method can be used in a number of ways to make nitrogen heterocycles and also carbocycles; various ring sizes are accessible.
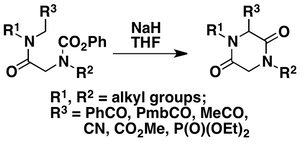
Formation of Piperazine-2,5-diones
Piperazine-2,5-diones, which are important structural motifs in several medicines, are formed by Dieckmann cyclization of substructures of the type R3CH2N(R1)C(O)-CH2N(R2)CO2Ph in which the terminal methylene (CH2) that is adjacent to nitrogen closes onto the carbonyl group of the phenyl carbamate unit at the other end of the chain. The starting materials are easily assembled by standard acylation and oxidation processes.
Synthesis of 1,3'-Bipyrroles

The 1,3'-bipyrrole system has recently been recognized as a subunit of a new class of important antibiotics that are extremely active against vancomycin-resistant bacteria. The method developed in this laboratory is the first general route to highly substituted 1,3'-bipyrroles and its use makes available numerous analogs for biological evaluation. Previously, the range of accessible analogs was severely limited, but the present route removes this restriction.
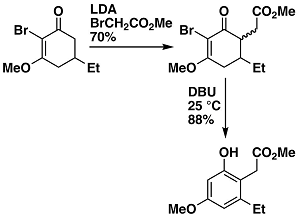
Synthesis of Alkyl-substituted Phenols
During work on the total synthesis of coleophomones A and B an unexpected observation lead to the development of a method for making substituted phenols. This method gives access to a wide range of aromatics appropriately functionalized for further elaboration in synthetic endeavors.
A Family of Routes to Substituted Phenols
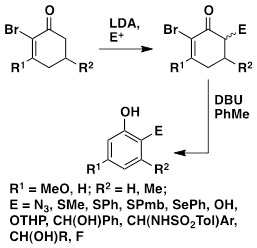
A new family of routes to substituted phenols has been developed. The sequence affords phenolic azides (ArN3), sulfides (ArSR, ArSAr'), selenides (ArSePh), alcohols [ArCH(OH)R], amino derivatives [ArCH(NHSO2-Ar')R], and 1,2-benzenediols. A complementary set of substitution patterns is obtained by DIBAL-H reduction or reaction with a Grignard reagent before aromatization; the latter process gives compounds in which the newly-introduced substituent is meta to the phenolic hydroxyl.
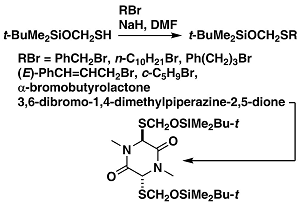
A Nucleophilic Protected H2S equivalent
t-BuMe2SiOCH2SH is a nucleophilic reagent for introduction of a protected bivalent sulfur; this reagent is complementary to our electrophilic reagent p-Me-C6H4-SO2SCH2OSiMe2Bu-t. The thiol reacts with alkyl bromides to give protected thiols. The protecting group can be removed with Bu4NF.
The Silicon Route to Cycloalkane-1.3-diones
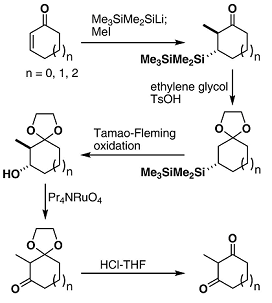
Conjugate addition of Me3SiMe2SiLi to cycloalk-2-en-1-ones, ketalization, Tamao-Fleming oxidation (Bu4NF, then H2O2, KHCO3), TPAP oxidation and acid hydrolysis generates 2-methyl cycloalkane-1,3-diones. Ketalization is needed in order to prevent addition of Me3Si- to the carbonyl. The pentamethyldisilanyl group has advantages over other silicon units that are used in Tamao-Fleming procedures.
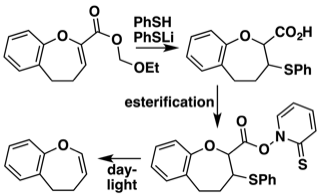
Radical decarboxylation route to hydrooxepine enol ethers
MPC1001 and related antibiotics contain a hydrogenated oxepine ring system. We have developed a mild approach to such compounds based on a photochemical decarboxylation. The starting enol ether esters are easily available. They readily undergo thiol addition and simultaneous deprotection of the carboxyl group. Formation of the Barton ester and exposure to daylight generates the oxepine system — all under very mild conditions suitable for synthesis of the MPC1001 antibiotic series.
Formation of meta-Aminophenols
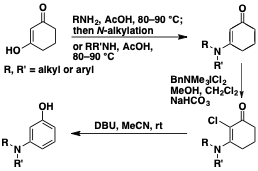
meta-Aminophenols are subunits of some agrochemicals and medicines and we have developed a mild route to such phenols. Cyclohexane-1,3-dione is readily converted into 3-aminocyclohex-2-en-1-ones in which the nitrogen carries two substituents, which are both aryl or alkyl units, or one of each type. Such compounds can be chlorinated at C(2) with the crystalline reagent BnNMe3.ICl2 and the resulting chloro enaminones undergo aromatization at room temperature on treatment with DBU in MeCN.
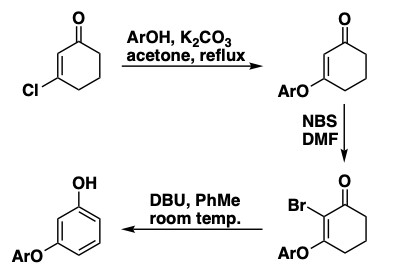
Formation of meta-(Aryloxy)phenols
The classical Ullmann reaction for making diaryl ethers uses a copper catalyst and a high temperature. In contrast, the aromatization step displayed on the right operates at room temperature and does not involve any heavy metals. By starting with a 3-chlorocyclohexenone, which is readily available in one step, all the problems of making a 3-substituted phenol which would be needed for the traditional Ullmann process are avoided.