Shuai Sun and Alex Brown
Department of Chemistry, University of Alberta, Edmonton, Alberta T6G 2G2, Canada
Abstract
Shuai Sun and Alex Brown
Department of Chemistry, University of Alberta, Edmonton, Alberta T6G 2G2, Canada
Abstract
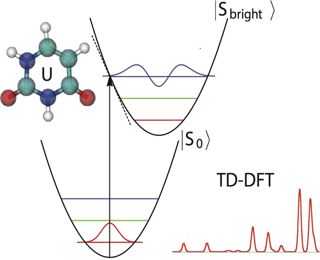
The resonance Raman spectrum of uracil is simulated using the Herzberg-Teller short-time dynamics formalism. The ground-state geometry is optimized at the levels of PBE0/aug-cc-pVTZ and B3LYP/aug-cc-pVTZ, respectively. The gradient of the bright excited state is computed using time-dependent density functional theory and spin-flip time-dependent density functional theory. The excited-state calculations are carried out in both the gas phase and implicit water using the conductor-like polarizable continuum model. The ground-state equilibrium structure is found to impact the resulting resonance Raman spectrum significantly. The simulated resonance Raman spectrum using the long-range corrected functionals, that is, CAMB3LYP and LC-BLYP, and based on the PBE0/aug-cc-pVTZ optimized ground-state structure shows better agreement with the experimental spectrum than using standard hybrid functionals, that is, PBE0 and B3LYP. The solvation effect leads to a change in the energetic order of the n → π* and π → π* excited states, and it improves the agreement with the experimental spectrum, especially with regard to the relative intensities of the peaks with frequencies greater than 1600 cm-1.
Return to Dr. Alex Brown's Publications
This page maintained by alex.brown@ualberta.ca of the Department of Chemistry, University of Alberta
Last updated September 8, 2014.