E. Pradhan, J.-L. Carreon-Macedo, J.E. Cuervo, M. Schroeder, and A. Brown
Department of Chemistry, University of Alberta, Edmonton, Alberta T6G 2G2, Canada
Abstract
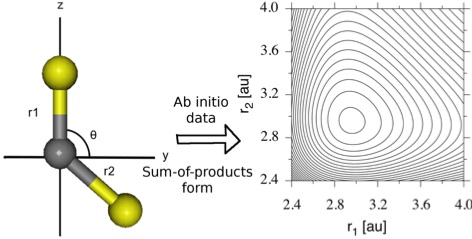
The ground state potential energy and dipole moment surfaces for CS2 have been determined at the CASPT2/C:cc-pVTZ, S:aug-cc-pV(T+d)Z level of theory. The potential energy surface has been fit to a sum-of-products form using the neural network method with exponential neurons. A generic interface between neural network potential energy surface fitting and the Heidelberg MCTDH software package is demonstrated. The potential energy surface has also been fit using the potfit procedure in MCTDH. For fits to the low-energy regions of the potential, the neural network method requires fewer parameters than potfit to achieve high accuracy - global fits are comparable between the two methods. Using these potential energy surfaces, the vibrational energies have been computed for the four most abundant CS2 isotopomers. These results are compared to experimental and previous theoretical data. The current potential energy surfaces are shown to accurately reproduce the low-lying vibrational energies within a few wavenumbers. Hence, the potential energy and dipole moments surfaces will be useful for future study on the control of quantum dynamics in CS2.
Return to Dr. Alex Brown's Publications
This page maintained by alex.brown@ualberta.ca of the Department of Chemistry, University of Alberta
Last updated August 13, 2013.